The clinical study was completed in July 2020.
Study title (Treatment period 1): A Phase I-II, Randomised, Double-Blind, Placebo Controlled, Safety and Tolerability Study of Intermittent Bilateral Intraputamenal Cerebral Dopamine Neurotrophic Factor (CDNF) Infusions Administered via an Investigational Drug Delivery System to Patients with Idiopathic Parkinson’s Disease (PD) of Moderate Severity.
Study title (Treatment Period 2): A Randomised, Double-Blind, Multi-centre, Active Treatment, Extension and Safety Study for Patients with Idiopathic Parkinson’s Disease (PD) Who Previously Completed the CDNF/DDS Main Study HP-CD-CL-2002.
Treatment with CDNF (Cerebral Dopamine Neurotrophic Factor) aims to modify the progression of Parkinson’s disease and to provide symptomatic relief. However, as this is a first-in-human study, the primary objectives are safety and tolerability.
Herantis Pharma Plc in collaboration with Renishaw Plc.
Main study protocol covering the Surgery and Treatment period 1:
The Treatment period 2 is being conducted under a separate Extension study protocol which also has been approved both in Finland and Sweden by the regulatory authorities and ethics committees.
The studies are registered at the ClinicalTrials.gov database with the numbers NCT03295786 and NCT03775538, and at the European Clinical Trials Database (EudraCT) with the numbers 2015-004175-73 and 2018-000346-19 accordingly.
Participation in the study is entirely voluntary. Anyone taking part in the study can change their mind at any time and withdraw from the study without providing a reason. Non-participation or withdrawal from the study will not affect current or future care of patients. If a patient chooses to discontinue participation in the study, he or she will be asked to complete the end-of-study procedures (e.g. final medical examination and laboratory tests) for their own safety.
Participation in the study may also be stopped at any time by the responsible doctor on medical grounds.
The Informed Consent Form, containing all relevant patient information, is available in Finnish and Swedish. The following versions for the Main study Informed Consent Form are currently available:
The following version for the Extension study Informed Consent Form is currently available:
The study drug is called CDNF (Cerebral Dopamine Neurotrophic Factor). It is a protein, which is found naturally in the body. In this study, its potential to prevent dopamine neuron degeneration and to stimulate neuron regeneration are being studied. As CDNF is a protein, it cannot be given as a pill or an injection as the body will not transport it to the brain. Therefore, in this study, CDNF is given using a drug delivery system, which has been implanted into the brain by a neurosurgeon.
CDNF has not been studied previously in humans. In the initial study, CDNF is compared to a placebo product in a 2:1 ratio. Patients are randomly assigned to either placebo, CDNF mid-dose or CDNF high-dose groups. After six months of treatment in the initial study (Treatment period 1), patients are offered to participate in an extension study in which all patients receive the active CDNF drug (Treatment period 2).
The CDNF drug is delivered to the brain via catheters, which are implanted by neurosurgeons in the putamen area of the brain. The catheters are connected to a port, which is fixed to the head, just behind the ear. The system also includes filters, which prevent bacteria and air from entering the brain.
This investigational drug delivery system has been previously implanted in more than forty patients in another clinical study.

The seventeen patients, who have been enrolled in the study have met the following criteria:
A patient’s eligibility to join the study has been assessed in detail during the screening phase at the start of the study.
Those patients who have not met the criteria for participation have been informed of the reasons, and they have continued to receive the same healthcare as they would normally receive.
On several visits, patients are asked not to take their Parkinson’s medication for at least ten hours prior to your appointment. Patients can take their usual medication as soon as the assessments have been completed.
Patients need to stay in the hospital or a hotel several times during the study, mostly one night but after the surgery for about four nights.
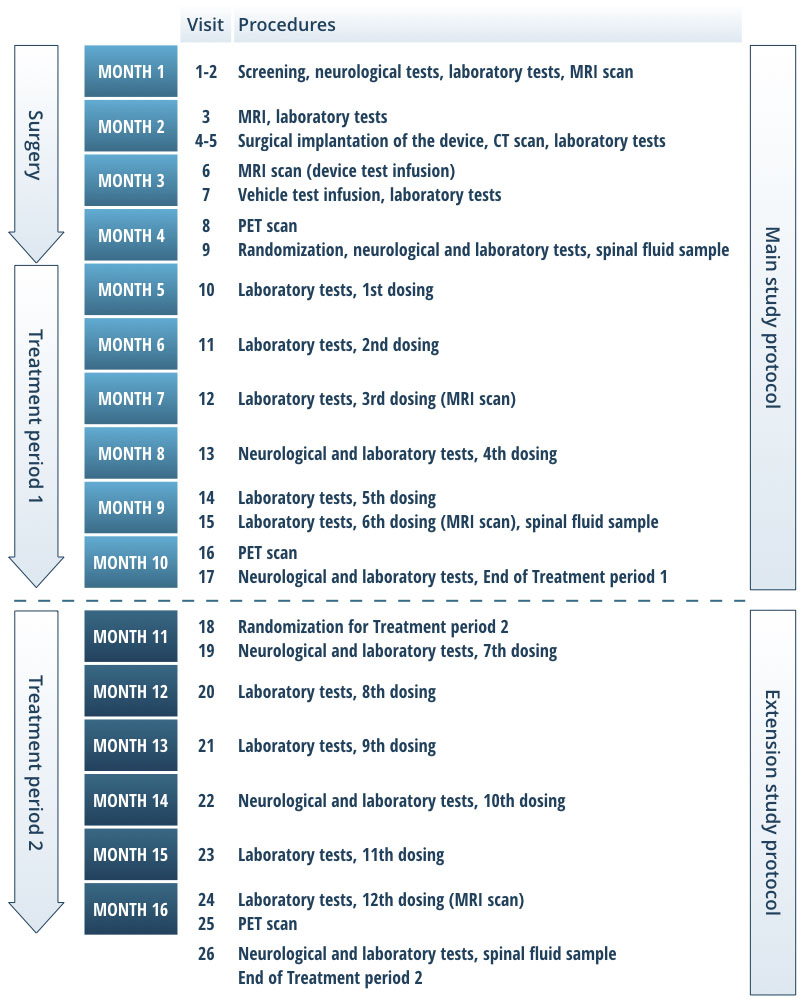
Figure 2. Study timeline and procedures per visit. The clinical study is composed of two studies performed under two different study protocols and patient consents. The first part of the study includes the device surgery and Treatment period 1. The second part includes Treatment period 2. After the Treatment period 2, the device will be closed from further access by removing the port. Alternatively, the entire device may be explanted.
The study doctor discusses with patients other available treatments, including their benefits and risks. Participation in the study does not influence the usual treatment of Parkinson’s and patients may continue with their current medication. However, treatment with other investigational study drugs or investigational medical devices is not permitted during this study. There are also medications that may not be used during this study. The study doctor discusses these medicines in detail with all participating patients.
The purpose of this study is to evaluate the safety and tolerability of CDNF and the implanted drug delivery system. A potential positive treatment effect on the symptoms of Parkinson’s disease will be evaluated, however, it is not known if the study patients will have any benefit from the treatment. Data from this study may in the future benefit people with Parkinson’s.
The study doctor will discuss the best care for patients in a timely manner before the end of the study. Patients will be informed about the options available for optimal treatment of their condition. The drug delivery device port behind the ear will be removed. The implanted drug delivery system will be left in place in the brain, unless otherwise decided by the study doctor or at the patient’s request.
The study doctor and the sponsor are obliged to inform the patients if any new information emerges during the study which could affect patients’ decision to take part or continue. The study doctor will discuss such information with the patients, and may possibly decide that it is best for a patient to withdraw from this study.
All study data are confidential and will be processed using only a code number. The study doctor will keep a code list in which the patients’ code numbers are linked to personal details, such as name and date of birth.
The study results may be published in medical journals and presented in scientific conferences, meetings and seminars. However, the identity of patients will not be revealed in any such report or presentation. For specific purposes, information may be transferred to regulatory authorities and to organizations cooperating with the study sponsor.
Descriptions of the Main and Extension studies are available in the ClinicalTrials.gov database (Main study NCT03295786, Extension study NCT03775538). This website will not have information that could identify the study participants but will include a brief summary of the study results. This site shows information only in English, but patients can contact the study staff at any time to ask about the publicly available information in patients’ own language.
If a patient decides to leave the study prematurely, the data that has been collected before the withdrawal of the consent will be used in the evaluation of the study.
Patients have the right to obtain once a year, free of charge, details of what personal information has been recorded about them, from where the information has been obtained and how it has been disclosed. Patients also have the right to request that any incorrect information is corrected. All patients are encouraged to talk to the study doctor if any erroneous information is noticed.
In order to confirm that the study has been duly conducted and to confirm the data collected about patients is correct, certain authorised people may have access to the medical records of the patients of the study. An authorised representative of the regulatory authority or the ethics committee may require access to the patients medical records in order to verify that the study has been duly carried out.
As in routine national healthcare, patients participating in this study are covered by the national patient insurance. The study sponsor has taken an insurance to cover the expenses of any damages that are considered to result from the patients’ participation in the study. Patients should contact their study doctor if they believe they have suffered an injury as a result of their participation in this study.
The hospital has received funding from the European Commission to perform this study. All patients’ participation costs will be covered, including travel to appointments and possible losses of income for the time spent at the study visits.



